
Case
A 41-year-old Gravida 3 Para 2002 presented for a routine anatomical survey at 20 weeks. Several anomalies were noted: an absent cavum septum pellucidum (Figure 1A), micrognathia (Figure 1B), abnormal pericallosal artery (Figure 1C), hypertelorism (Figure 1D), nuchal fold thickening (Figure 1E), a complex cardiac defect (Figure 1F and 1G), and an absent left kidney (Figure 1H), Additional findings are listed in Table 1A. Amniocentesis revealed an abnormal male karyotype: Trisomy 22 (47, XY +22) (T22) (Table 1B). Microarray detected mosaic gain, around 80%, of all markers on chromosome 22 (Table 1B). Amniotic fluid alpha-fetoprotein was analyzed and found to be within normal range. Fetal echocardiogram identified a double outflow from the left ventricle, a large ventricular septal defect, and a possible mispositioning of the great arteries without alterations to flow throughout the ventricles or the great vessels.
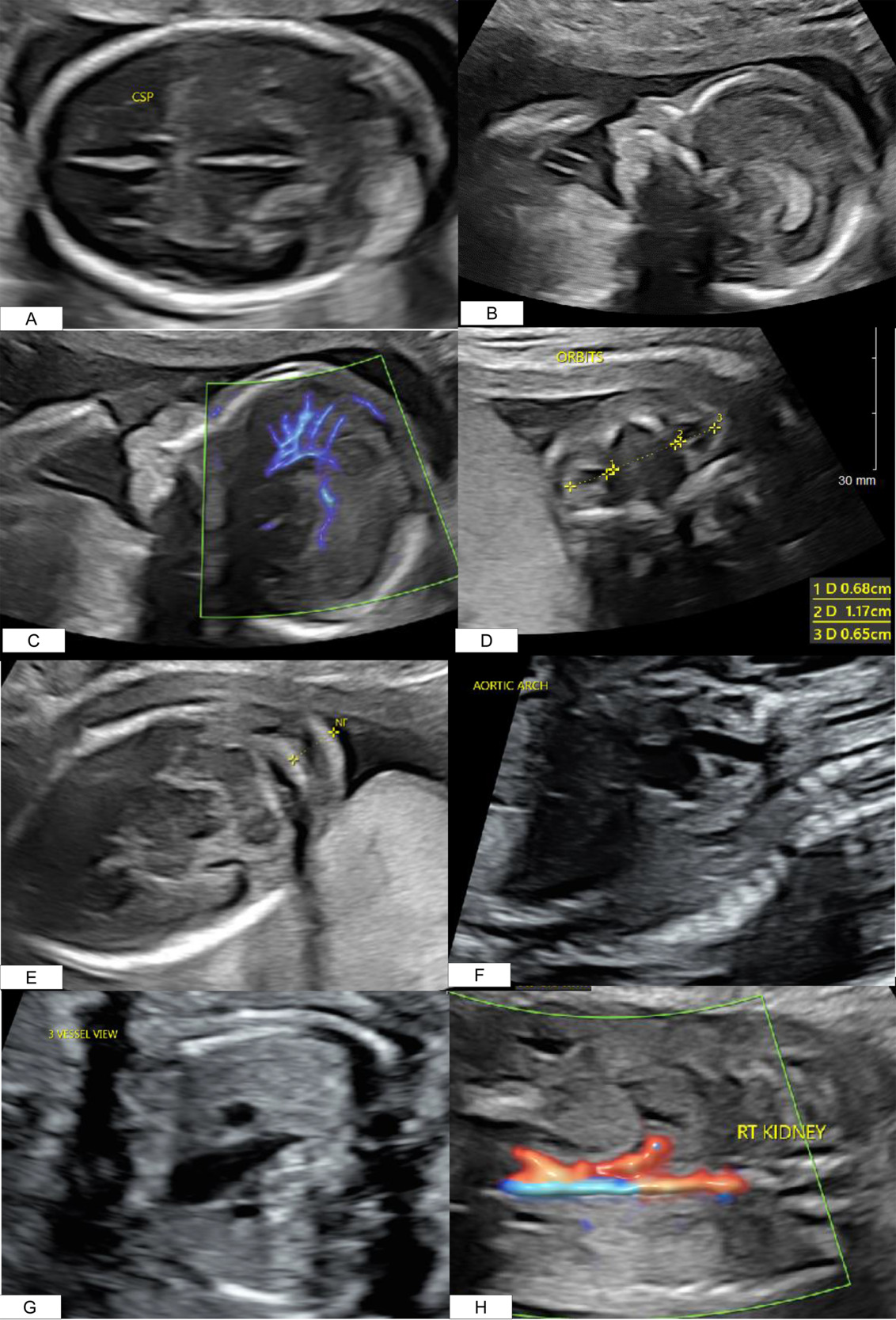
At 37 weeks and 0 days, the infant was delivered by a modified Ex-utero Intrapartum Treatment Procedure for palliative indications. Multiple intubation attempts were made and ultimately, the infant required emergent tracheostomy placement and positive pressure ventilation. His birth weight was 1730g (<1st percentile), and APGARs were 1,2,2,2,2,5. On physical exam, the neonate had multiple anomalies (Table 1A). On auscultation of the chest, decreased breath sounds were noted bilaterally, a holo-systolic III/VI murmur was heard, pulses were strong in all extremities, no cry or response to tactile or noxious stimuli was elicited, and hypertonicity was noted in all extremities. On exam of the face and head, the following findings were noted: microcephaly, micrognathia with retrognathia, cleft palate, absent left eye, low-set ears with ear crease and pit, and wide-set eyes. No integumentary, abdominal, or genitourinary anomalies were noted.
An initial bedside echocardiogram showed a large coronary sinus, two separate atrioventricular valves, a severely depressed ejection fraction (15-20%), a double outlet right ventricle, a small pericardial effusion, and moderate tricuspid regurgitation. Bilateral chest tubes were placed due to bilateral pneumothoraxes, and a repeat echocardiogram was performed, which demonstrated the following additional findings: a hypoplastic ascending, and transverse aortic arch with possible interruption, as well as a large patent ductus arteriosus with right to left shunt. On abdominal ultrasound, no definite renal tissue was identified. The right adrenal gland appeared normal, but the left was not visualized. The liver, gallbladder, pancreas, spleen, aorta, and inferior vena cava appeared normal. On day of life one, the parents elected to withdraw care.
The placenta had diffuse distal villous dysmaturity, the umbilical cord had a single umbilical artery, a recent retroplacental hematoma was noted and <5% of the placenta had remote infarcts.
Autopsy identified microcephaly, cleft palate, micrognathia with retrognathia, left anopthalmos with absent ophthalmic nerve, absent corpus callosum, low set ears, short bilateral radii and femurs, double outlet right ventricle with left ventricular hypoplasia and ventricular septal defect, a minuscule right kidney and an absent left kidney, Potter sequence with pulmonary hypoplasia and alveolar wall thickening.
Discussion
T22 is traditionally a lethal anomaly that results in in-utero demise. Due to the mosaic nature of this case, the fetus reached term gestational age and was able to be resuscitated. A recent systematic review of the literature has identified 25 postnatal and 18 prenatal cases reported in the literature (Trevisan, Meroni, Leoni, et al. 2024). There are few reports of liveborn infants with this condition, and fewer that describe liveborn infants with a high level of mosaicism (the highest reported in a liveborn neonate was 35%) (Trevisan, Meroni, Leoni, et al. 2024; Abdelgadir, Nowaczyk, and Li 2013; Wang et al. 2007; Basaran, Berkil, Ay, et al. 2001; Stiefmeier et al. 2004; Chen, Huang, Chern, et al. 2019). Our case report describes a liveborn infant with a high level of mosaicism (80%) and is the first to demonstrate anopthalmos with an absent ophthalmic nerve, absent corpus callosum, and renal agenesis.
Authorship Contributor Statement
ER: Direct patient care, literature review, manuscript writing; KN: literature review, direct patient care, CB: manuscript writing direct patient care; VB: direct patient care, figure development, manuscript editing; SD: direct patient care, manuscript editing, provided expert clinical guidance; EM: direct patient care, manuscript editing, provided expert clinical guidance; TT: direct patient care, provided expert clinical guidance, reviewed final manuscript
Conflict of interest statement
The authors report no conflict of interest in the conduct and reporting of this study.
Funding statement
No funding source was used in study design, analysis of data, interpretation of results, or writing of the report
The ethics approval statement is not applicable. Patient provided consent for publication of this case report regarding their
Data availability statement is not applicable.